aMD简介
在分子动力学模拟中,精准的力场和高效的采样始终是研究的热点问题。aMD(accelerated molecular dynamics,加速分子动力学)便是由UCSD的McCammon group提出的一种增强采样的方案。
aMD是一种通过填平势能面实现的增强采样技术,当体系的势能小于某个参考值的时候为体系加上一个非负的提升势。提升势降低了反应的能垒,因而能够加速体系在不同的低能状态之间的转换,从而采样到不同的构象状态,观测到常规MD中难以看到的跨能垒事件。
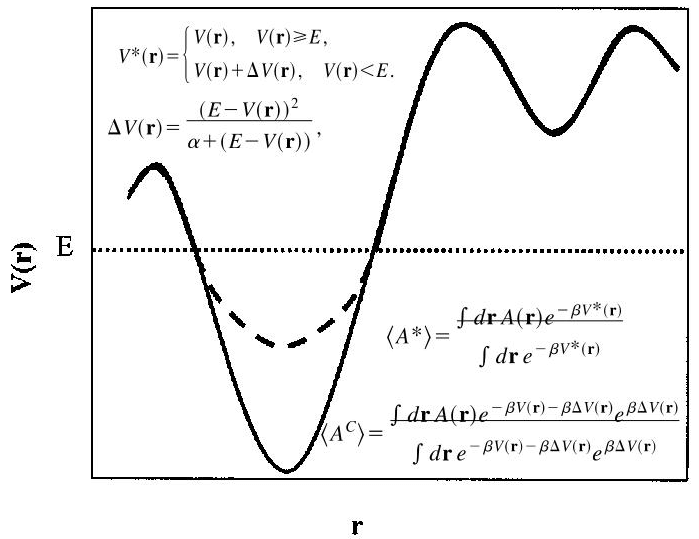
如图所示,为了让体系能够逃离势能的极小值,当体系的势能\(V(r)\)低于某个固定值 \(E\) 时,aMD会自动添加一个提高势\(\Delta\left( V\left( r\right) \right)\)使得体系更容易跨过能垒,从而能够加速采样。
通过观察系综平均的计算公式可以发现,原体系的系综平均 \(\langle A\rangle\) 与增强采样之后的系综平均\(\langle A^{\ast}\rangle\)只是相差了一个因子\(e^{\beta\Delta\left( V\left( r\right) \right)}\),故只需对每一个构象乘以相应的因子,得到的 \(\langle A^C\rangle\) 与原体系的系综平均 \(\langle A\rangle\) 相同。如此以来便可通过reweighting由增强采样的自由能得到常规MD的自由能描述,进而计算其它热力学性质。
需要注意的是,这里我们提到的势能\(V(r)\)一般只考虑体系的二面角项和非成键项对应的能量,而不是体系所有的能量项之和(Amber中可以通过iamd选项来决定考虑体系的二面角
or 总势能)。
从\(\Delta\left( V\left( r\right) \right)\)的公式中可知,\(E\)和\(\alpha\)是两个需要调整的参数。若\(\alpha=0\),则相当于将势能固定在\(E\),变成随机游走;若\(\alpha\)值太大,则?
关于\(E\)的选择,若\(E\)太小则\(V(r)>E\)始终成立,相当于没有增强采样。故而,在aMD之前还需要进行一段常规MD,然后将E取为势能的平均值。
通常,aMD的加速效果能将模拟的时间缩短一个数量级。