Amber中使用aMD
传统的分子动力学模拟时间尺度一般在数百纳秒,难以捕捉到动辄在微秒甚至毫秒尺度说方能观测到的生物过程。本教程便是通过在Amber中使用aMD来研究BPTI在微秒尺度上的构象转变,并与DE Shaw发表的毫秒尺度的常规动力学进行比较。英文版教程来源于Amber官网。
aMD简介
aMD通过修改势能来降低局部能垒的高度,从而加速采样。aMD降低能垒有两种方式,分别表现为势能面上的向下填坑和削减峰值(raising minima and lowering barriers )。前者使用更为广泛,也是本教程中使用的方案(两种方式均被Amber支持)。
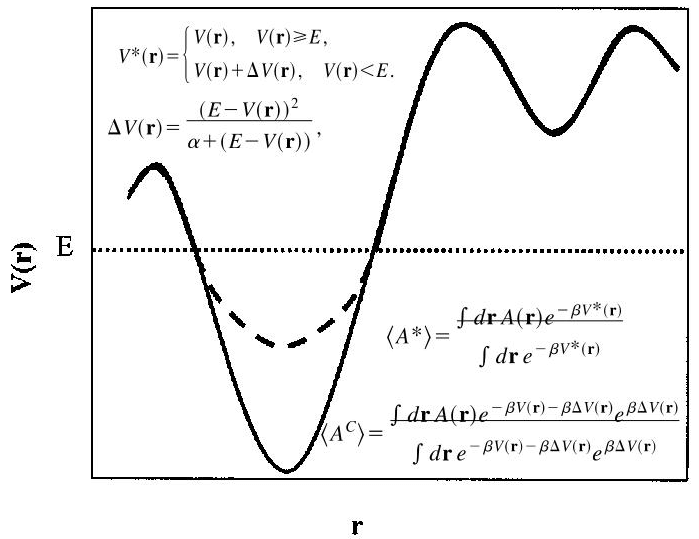
由上图可知,aMD修改势能需要修改E和a两个参数。
Amber支持以三种方式修改势能,分别为仅对torsional对应的势能项添加提高势(iamd=2);对整个势能项添加提高势(iamd=1);对二者同时添加提高势(iamd=3),于是对应如下四个参数:
1
2
3
4a) EthreshP. Average total potential energy threshold.
b) alphaP. Inverse strength boost factor for the total potential energy.
c) EthreshD. Average dihedral energy threshold.
d) alphaD. Inverse strength boost factor for the dihedral energy.
模拟步骤
Generating and Relaxing the Initial Structure
- 修改晶体结构(5PTI)中的氢(DtoH),并移除多余的水分子,得到 5PTI-DtoH-dry.pdb。
- 形成二硫键然后将体系溶解在TIP4P-Ew水中,使用ff99SB-I力场(模拟DE
Shaw文章中的protocol)。 脚本如下(处理好的文件见教程中的链接):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15$AMBERHOME/bin/tleap
> source ~/amber11intel/dat/leap/cmd/leaprc.ff99SBi
> loadOff solvents.lib
> loadOff tip4pbox.off
> loadOff tip4pewbox.off
> loadAmberParams ~/amber11intel/dat/leap/parm/frcmod.tip4pew
> HOH=TP4
> mol = loadpdb 5PTI-DtoH-dry.pdb
> bond mol.55.SG mol.5.SG
> bond mol.30.SG mol.51.SG
> bond mol.14.SG mol.38.SG
> addions2 mol Cl- 6
> solvatebox mol TIP4PEWBOX 10.5
> saveamberparm mol bpti-ff99SBi-large.prmtop bpti-ff99SBi-large.inpcrd
> quit - 运行MD 分为如下步骤,需要的输入文件见文末。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
201. Minimize only the water, restraining the protein (20000 cycles)
2. Let water move (NTP, 300K), restraining the protein
3. Minimize water and protein (20000 cycles)
4. Heat the system, restraining the protein (NVT 0 to 300K)
5. Relax the system, restraining the protein heavy atoms (NPT, 300K, 0.5ns)
6. Relaxthe system (NPT, 300K, 5ns)
在6个子文件夹[1-6]_中分别进行:
$AMBERHOME/bin/pmemd.cuda -O -i ../min_wat.in -o min_wat.out -p ../*.prmtop -c ../*.inpcrd -r min_wat.rst -ref ../*.inpcrd
$AMBERHOME/bin/pmemd.cuda -O -i ../md_wat.in -o md_wat.out -p ../*.prmtop -c ../1_/*.rst -r md_wat.rst -ref ../1_/*.rst
$AMBERHOME/bin/pmemd.cuda -O -i ../min_sys.in -o sys_min.out -p ../*.prmtop -c ../2_/*.rst -r sys_min.rst -ref ../2_/*.rst
$AMBERHOME/bin/pmemd.cuda -O -i ../heat.in -o heat.out -p ../*.prmtop -c ../3_/*.rst -r heat.rst -ref ../3_/*.rst
$AMBERHOME/bin/pmemd.cuda -O -i ../density.in -o density.out -p ../*.prmtop -c ../4_/*.rst -r density.rst -ref ../4_/*.rst
$AMBERHOME/bin/pmemd.cuda -O -i ../eq.in -o eq.out -p ../*.prmtop -c ../5_/density.rst -r eq.rst
Running the aMD calculations and data collection.
通过上一步我们已经获得平衡之后的结构eq.rst,接下来通过运行一段短的常规MD来获取aMD所需的参数。
通过常规MD我们得知体系的总势能平均值为-47128
kcal/mol,二面角势能项(dihedral energy)平均值为595
kcal/mol。考虑到BPTI具有58个残基,并且整个体系含有18226个原子,我们通过如下公式计算aMD的参数:
1
2
3
4
5
6
7a) EthreshP: E(tot)= -47128 kcal/mol + (0.16kcal/mol/atom * 18226 atoms) = -44212 kcal/mol
b) alphaP: Alpha(tot)= (0.16 kcal/mol/atom * 18226 atoms) = 2916 kcal/mol # Amber手册中为 0.15-0.19 kcal/mol/atom
c) EthreshD: E(dih)=595 kcal/mol + (4 kcal/mol/residues * 58 solute residues) = 827 kcal/mol
d) alphaD: Alpha(dih)=(1/5)*(4 kcal/mol/residues * 58 solute residues) = 46.4 kcal/mol # Amber手册中为 3.5 kcal/mol/residues
现在进行500ns的aMD模拟(GTX690 单个GPU需要52小时) 1
$AMBERHOME/bin/pmemd.cuda -O -i amd.in -o amd.out -p ../*.prmtop -c ../6_/eq.rst -r prod.nc
aMD结果处理
PCA compared with DE Shaw's result
以下是PCA分析的结果,将不同轨迹投影到500ns
aMD获得的前两个PC上。其中,右图是500ns aMD轨迹的PCA,中间是500ns
常规MD(与aMD相同参数)投影到aMD获得的PCA空间,左图是DE
Shaw的轨迹投影到aMD获得的PCA空间。 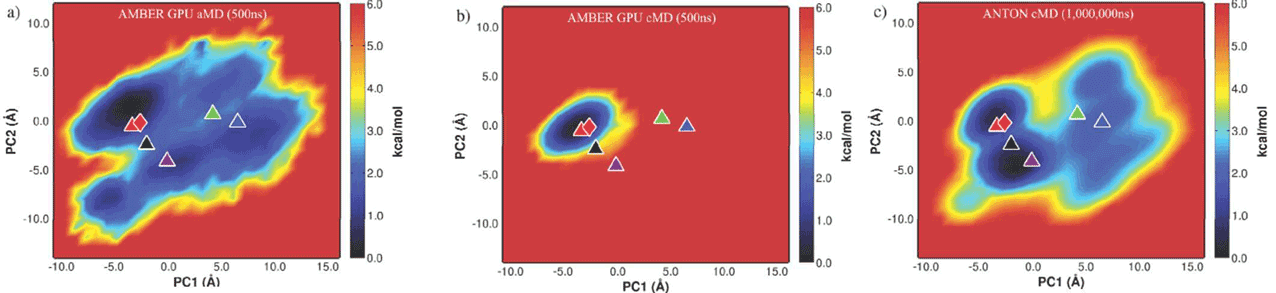
由此可见通过相对短时间的aMD模拟便能够采样到与DE Shaw长时间模拟等价的相空间。
产生aMD的PCA向量并将轨迹投影到PCA空间使用的cpptraj脚本如下(运行失败,未继续尝试):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23cat > ptraj.in
# Usage: cpptraj < ptraj.in > ptraj.out
parm ../bpti-ff99SBi.prmtop
trajin ../7_/mdcrd 1 last 1
reference ../bpti-ff99SBi.inpcrd
center origin :1-58
image origin center
strip :WAT,Cl-
rms reference mass :1-58@CA,C,N out AMD/bpti/pca-analysis/PDBfit/RMSD.out
matrix covar name matrixdat @CA out covmat-ca.dat
analyze matrix matrixdat out evecs-ca.dat vecs 174
analyze matrix matrixdat name evecs-ca vecs 174
analyze modes fluct out analyzemodesflu.dat stack evecs-ca beg 1 end 174
analyze modes displ out analyzemodesdis.dat stack evecs-ca beg 1 end 174
projection modes evecs-ca.dat out pca12-ca beg 1 end 3 @CA
go
类似地,将500ns常规MD,或者DE Shaw的轨迹进行投影: 1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22# align 轨迹,并保存为binpos格式
reference ../scratch.md99SBi/av-CA.pdb
trajin shaw1-CA.pdb
trajin shaw2-CA.pdb
trajin shaw3-CA.pdb
trajin shaw4-CA.pdb
trajin shaw5-CA.pdb
trajin xray-CA.pdb
trajin BPTI-SHAW/DESRES-Trajectory-bpti-c-alpha/bpti-c-alpha/bpti-c-alpha-000.dcd 1 -1 2
...
trajin BPTI-SHAW/DESRES-Trajectory-bpti-c-alpha/bpti-c-alpha/bpti-c-alpha-041.dcd 1 -1 2
rms reference out rms-av.dat :4-54@CA
trajout alav-CA.bps binpos nobox
go
# 投影到之前产生的PCA空间
trajin alav-CA.bps
projection modes ../scratch.aMD-C67-NEW-first500ns/evecs-ca.dat out pca12-ca beg 1 end 3 :4-54@CA
go
Reweighting the aMD results
aMD
的一个主要优势便是它能够将模拟的分布reweight成未受扰动的常规MD。在aMD过程中会产生amd.log文件(可通过-amdlog选项指定),包含进行reweighting
所需要的所有信息,它的输出频率与轨迹文件的输出频率一致,每一行表示一帧,内容如下:
1
2
3#All energy terms stored in units of kcal/mol
#ntwx,total_nstep,Unboosted-Potential-Energy, Unboosted-Dihedral-Energy,Total-Force-Weight,Dihedral-Force-Weight,Boost-Energy-Potential,Boost-Energy-Dihedral
及其对应的描述: 1
2
3
4
5
6
7
8
9
10
11a) Unboosted-Potential-Energy = Total Potential Energy without boost added, kcal/mol
b) Unboosted-Dihedral-Energy = dihedral energy without boost added, kcal,mol
c) Total-Force-Weight = The force scaling factor calculated from the boost to the Total Potential Energy
d) Dihedral-Force-Weight = The dihedral force scaling factor from dihedral boost
e) Boost-Energy-Potential = The boost energy in kcal/mol
f) Boost-Energy-Dihedral = The dihedral boost energy in kcal/mol
NOTE: Amber14之前的版本里,amd.log中最后两列boost energy 的单位是 KT,从Amber14之后单位改为 kcal/mol.
对aMD的结果进行reweighting,可以关注另一个教程,其中提供了方便的python脚本,能够进行一维和二维(比如PCA,Phi-Psi分布)的reweighting,这里同时包含一个C语言版本的脚本
reweight-KDE.c,利用Kernel Density Estimation进行二维aMD
reweighting。
这里以如丙氨酸二肽的Phi_Psi分布为例进行简单介绍,进行reweighting之前我们需要得到每一帧结构对应的Phi_Psi:
1
2
3
4
5
6
7
8
9
10
11
12# cpptraj
# 2 ALA PHI: (1 ACE C)-(2 ALA N)-(2 ALA CA)-(2 ALA C) -180 -180
# dihedral iat 5, 7, 9, 15,
dihedral phi :1@C :2@N :2@CA :2@C out Phi_Psi
# 2 ALA PSI: (2 ALA N)-(2 ALA CA)-(2 ALA C)-(3 NME N) -180 -180
# dihedral iat 7, 9, 15, 17
dihedral psi :2@N :2@CA :2@C :3@N out Phi_Psi
go1
2
3
4
5
6
7# For AMBER14:
# awk 'NR%1==0' amd.log | awk '{print ($8+$7)/(0.001987*300)" " $2 " " ($8+$7)}' > weights.dat
# For AMBER12:
# awk 'NR%1==0' amd.log | awk '{print ($8+$7)" " $3 " " ($8+$7)*(0.001987*300)}' > weights.dat
若使用Python代码: 1
python PyReweighting-2D.py -input Phi_Psi -Emax 100 -discX 6 -discY 6 -job amdweight_MC -order 10 -weight weights.dat | tee -a reweight_variable.log
1
2
3gcc reweight-hammelberg.c -o reweight-hammelberg -lm # 先进行编译
./reweight-KDE
# 运行MD所需的in文件如下:
|